Abstract
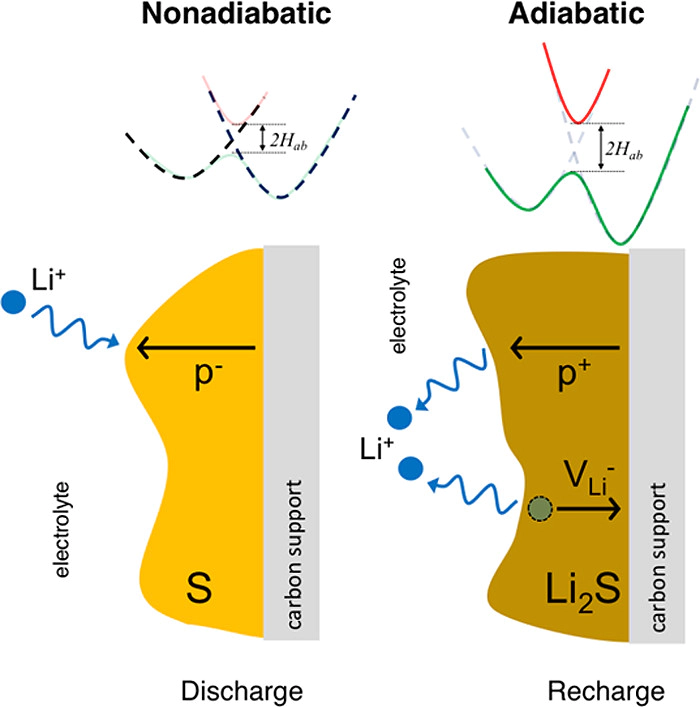
The insulating nature of the redox end members in Li–S batteries, α-S and Li2S, has the potential to limit the capacity and efficiency of this emerging energy storage system. Nevertheless, the mechanisms responsible for ionic and electronic transport in these materials remain a matter of debate. The present study clarifies these mechanisms—in both the adiabatic and nonadiabatic charge transfer regimes—by employing a combination of hybrid-functional-based and constrained density functional theory calculations. Charge transfer in Li2S is predicted to be adiabatic, and thus is well-described by conventional DFT methodologies. In sulfur, however, transitions between S8 rings are nonadiabatic. In this case, conventional DFT overestimates charge transfer rates by up to 2 orders of magnitude. Delocalized holes, and to a lesser extent, localized electron polarons, are predicted to be the most mobile electronic charge carriers in α-S; in Li2S, hole polarons dominate. Although all carriers exhibit extremely low equilibrium concentrations, and thus yield negligible contributions to the conductivity, their mobilities are sufficient to enable the sulfur loading targets necessary for high energy densities. Our results highlight the value of methods capable of capturing nonadiabaticity, such as constrained DFT. These techniques are especially important for molecular crystals such as α-S, where longer-range charge transfer events are expected. Combining the present computational results with prior experimental studies, we conclude that low equilibrium carrier concentrations are responsible for sluggish charge transport in α-S and Li2S. Thus, a potential strategy for improving the performance of Li–S batteries is to increase the concentrations of holes in these redox end members.