Abstract
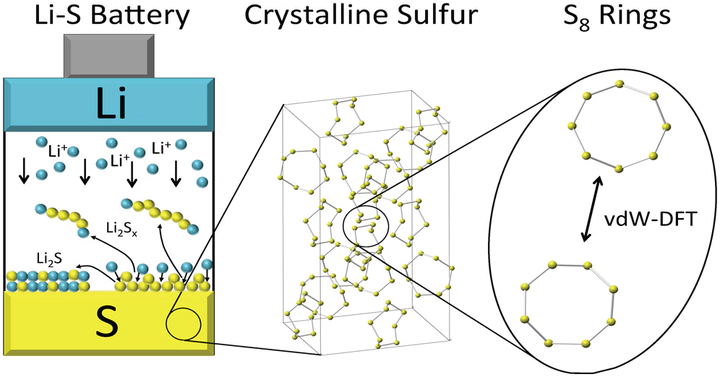
The properties of the solid-phase redox end members, α-S, β-S, Li2S, and Li2S2, are expected to strongly influence the performance of lithium–sulfur batteries. Nevertheless, the fundamental thermodynamic and electronic properties of these phases remain poorly understood. From a computational standpoint, the absence of these data can be explained by the omission of long-ranged van der Waals interactions in conventional density functionals; these interactions are essential for describing the molecular-crystal nature of S-based compounds. Here we apply van der Waals augmented density functional theory (vdW-DF), quasi-particle methods (G0W0), and continuum solvation techniques to predict several structural, thermodynamic, spectroscopic, electronic, and surface characteristics of these phases. The stability of the α allotrope of sulfur at low temperatures is confirmed by calculating the sulfur phase diagram. Similarly, the stability of lithium persulfide, Li2S2, a compound whose presence may limit capacity, was assessed by comparing the energies of several hypothetical A2B2 crystal structures. In all cases Li2S2 is predicted to be unstable with respect to a two-phase mixture of Li2S and α-S, suggesting that Li2S2 is a metastable phase. Regarding surface properties, the stable surfaces and equilibrium crystallite shapes of Li2S and α-S were predicted in the presence and absence of a continuum solvation field intended to mimic the effect of a dimethoxyethane (DME)-based electrolyte. In the case of Li2S, the equilibrium crystallites are comprised entirely of stoichiometric (111) surfaces, while for α-S a complex mixture of several facets is predicted. Finally, G0W0 calculations reveal that all of α-S, β-S, Li2S, and Li2S2 are insulators with band gaps greater than 2.5 eV.